


By Nat Kolber, scientist-in-residence at Civilization Ventures, with contributions from the whole Civilization Ventures team
Contents:
The transformative promise and current state of genetic medicines
Limitations of adeno-associated viruses (AAVs) and lentivirus
The New Frontier: our investment in Nanite and its polymer nanoparticles (PNPs)
The transformative promise and current state of genetic medicines
Genetic medicines have the potential to treat and even cure diseases that are beyond the reach of small molecule or biologic drugs. The past decade has seen a proliferation of tools for editing DNA and RNA, upregulating and downregulating gene expression, influencing mRNA splicing, and engineering immune cells for the treatment of cancers and autoimmunity. Combined with new insights into the molecular drivers of disease, these tools are paving the way for more precise, effective, and personalized treatments for both common and rare genetic disorders1.
At Civilization Ventures, we aim to build on these advancements to vastly expand the scope of genetic medicines. We imagine treating and curing rare genetic diseases that affect any tissue in the body, harnessing immune cells with sophisticated logic to precisely target and eliminate cancer, and engineering living therapies that integrate seamlessly into the body, continuously sensing, adapting, and responding to disease in real time. This vision is becoming reality through pioneering companies like Nanite, our bet on delivery for next generation medicines, whose innovative polymer nanoparticle delivery platform and AI-driven approach to formulation discovery exemplify the breakthrough thinking needed to overcome current limitations in gene delivery. With such advances, we can envision genetic medicines that restore, regenerate, and rejuvenate damaged and aging tissues. We believe in a future where this can be done safely, cost-effectively, and at scale.
Historically, the promise of genetic medicines has been severely limited by our ability to deliver genetic information to cells. The 23 FDA-approved genetic medicines2 make the need for new delivery technologies obvious:
*conditional approval
Limitations of adeno-associated viruses (AAVs) and lentivirus
Approved genetic medicines mostly rely on one of two delivery modalities: adeno-associated virus (AAV) and lentivirus. These are viruses engineered to express a gene of interest (a transgene) and to be non-replicating (i.e., unlike viruses found in nature, they are unable to spread from one cell to another).
AAV is a widely used delivery vehicle because it is non-pathogenic and certain naturally occurring as well as engineered variants (serotypes) have an affinity for specific cell types. After infecting a cell, the AAV genome is expressed as an episome, a piece of DNA that is separate from the cell’s genome. Since episomes are not replicated during cell division, the therapeutic product of the episomal expression of an AAV is less effective in rapidly dividing cells unless used in conjunction with a gene-editing tool like CRISPR-Cas.
In contrast, lentivirus is a modified version of human immunodeficiency virus (HIV) that takes advantage of HIV’s natural ability to integrate into the host’s genome. This allows the introduced transgene to remain stable and active even in dividing cells. However, lentivirus’ random insertion into the genome poses the risk of insertional mutagenesis,* i.e., cancer caused by the disruption of a cancer-suppressing gene3.
Both modalities suffer from four critical liabilities:
Cargo Capacity. Viral delivery vehicles have characteristic cargo capacities defined by the virus’ structure and size of its genome. Adeno-associated viruses can only package genes ~4,700 nucleotides long (4.7 kb), while lentivirus has a packaging capacity of up to ~12,000 nucleotides (12 kb). Many genes of therapeutic interest exceed the packaging capacity of AAV, especially when accounting for regulatory elements that are needed for gene expression: for example, ABCA4 (for the treatment of Stargardt disease) is ~6,800 nucleotides long and CEP290 (for the treatment of Leber congenital amaurosis) is ~7,400 nucleotides long. Certain genes even exceed the packaging capacity of lentivirus, like dystrophin (for the treatment of Duchenne muscular dystrophy) at over 11,000 nucleotides long. Towards overcoming this limitation, CV portfolio company Tacit Therapeutics has developed splicing-directed repair technology that can repair large genes that would otherwise exceed the packaging capacity of viral vectors. However, packaging many genes spanning tens of thousands of nucleotides will be necessary for engineering smart cells that autonomously sense and respond to disease, correcting complex multifactorial disorders, regenerating entire tissues, and ultimately developing living medicines that can dynamically evolve alongside their human hosts.
Immunogenicity + toxicity. The human immune system evolved to recognize and destroy viral proteins. For some applications, a strong immune response is desired: for example, CV portfolio company Siren Biotechnology is developing AAV-based gene therapies that stimulate the immune system to attack cancer cells. For many applications, however, viral vector-based gene therapies must be co-administered with broadly immunosuppressive drugs that can leave patients vulnerable to infection and are associated with their own toxicities. Even then, the immune system can blunt the efficacy of gene therapies and cause massive and sometimes deadly immune reactions*.
While less immunogenic vectors like AAV can be effective upon initial dosing, the immune system “learns” to recognize the viral capsid after the initial exposure, precluding repeated dosing that would be necessary to titrate dosing or deliver a future gene therapy4. Most problematically, it is very difficult to predict immunogenicity before a gene therapy enters clinical trials.Biodistribution + tissue targetability. Both viral and non-viral vectors tend to accumulate in the liver. This has paved the way for many innovative therapies: for instance, CV portfolio company Excision Biotherapeutics is leveraging CRISPR-Cas to precisely cut hepatitis B DNA from liver cells, aiming to cure a disease that otherwise can persist for a lifetime. However, despite efforts to target other tissues, availability in non-liver tissues is often too low for therapeutic efficacy. This has limited the development of treatments targeting the brain, muscle, bone marrow, and many other tissues.5 Achieving therapeutic doses in extrahepatic tissues can require large doses of virus, driving up cost of goods and toxicity*.
Even when vectors successfully reach extrahepatic tissues, they can still accumulate in the liver, leading to serious side effects. A notable example is Novartis’ Zolgensma for spinal muscular atrophy, which has been associated with liver toxicity and patient deaths due to the buildup of viral capsids in the liver. Aside from the delivery vehicle, off-tissue expression of the transgene presents additional risks, including increased chances for off-target edits by gene editors, immune reactions to the transgene, the disruption of normal functions in non-target cells, and the potential for germline expression (which could be inherited by future generations with unpredictable consequences).Manufacturing and cost. Manufacturing viral gene therapies is more challenging than even biologics, as viruses are more complex, use less well-established production systems, and require more challenging processes for purification and characterization. This process is extremely cost-intensive, bringing cost-of-goods to ~$100k-1MM+ per dose. Moreover, economies of scale in viral vector manufacturing fail for both the small number of doses needed to address ultra-rare diseases (where low sales don’t justify the fixed cost of production) and the large number needed to treat common neurodegenerative and metabolic disease (where production and downstream purification processes cannot be efficiently scaled up).
These limitations have led to the concentration of FDA-approved genetic medicines in a narrow range of disorders. Many of these drugs involve modified blood cells, including six CAR- and one TCR-T treatments for blood cancers, two therapies for sickle cell disease, and two for ß-thalassemia. This is because blood cells are particularly amenable to genetic engineering ex vivo: they can be readily isolated, engineered to express a transgene, then reintroduced into the patient. This approach circumvents challenges related to immunogenicity and tissue-specific targeting, permitting the use of randomly integrating and more immunogenic viral vectors like lentivirus. Similarly, the scope of diseases treatable with AAV-based therapies is constrained by the virus’ limited cargo capacity and issues with biodistribution: most are either delivered locally or target the liver, including the two gene therapies approved for hemophilia B.
Emerging technologies for gene delivery
Several emerging technologies seek to address these issues and expand genetic medicines to new indications. As described below, however, each of these approaches also comes with its own liabilities:
Lipid nanoparticles (LNPs). You may recognize LNPs from their pivotal role in mRNA-based COVID-19 vaccines. These non-viral gene delivery vehicles consist of a lipid (fat) mixture surrounding mRNA or DNA. While LNPs can provoke an immune response, they are considered less immunogenic than viral vectors and are also significantly cheaper to manufacture. Unlike viral vectors, LNPs do not have a strict limit on cargo size and have been used to deliver larger genes like Cas9, although very large cargoes could lead to reduced packaging and delivery efficiency.
Due to their tendency to accumulate in the liver, most LNP-based gene therapies in clinical trials are either delivered locally or target the liver. Modifying lipid composition ** or adding proteins that target specific cell receptors* could enable extrahepatic delivery. However, these modifications may increase manufacturing costs and the risk of immunogenicity. To address this challenge, our portfolio company Kernal Bio has engineered tumor-specific and immune-specific LNPs capable of selective delivery to target cells, minimizing off-target liver accumulation and immunogenicity while improving overall efficacy.
LNPs enter cells in a process called endocytosis, wherein cells engulf extracellular materials within a lipid bilayer to form endosomes. Endosomes acidify and break down their contents as they mature. For successful gene delivery, LNPs must break apart and escape the endosome before this happens. However, this process is highly inefficient, resulting in only a small fraction of LNPs successfully delivering their cargo. Moreover, component lipids can trigger inflammation while the process of endosome disruption itself can cause inflammation and toxicity, as seen in the Phase I failure of Verve Therapeutics’ PCSK9 base editor for the treatment of hypercholesteremia*.
Finally, LNPs are not well-suited to delivering DNA to non-dividing cell types as they lack mechanisms to enter the nucleus. In contrast, viruses have evolved more efficient strategies to enter cells, escape the endosome, and enter the nucleus.
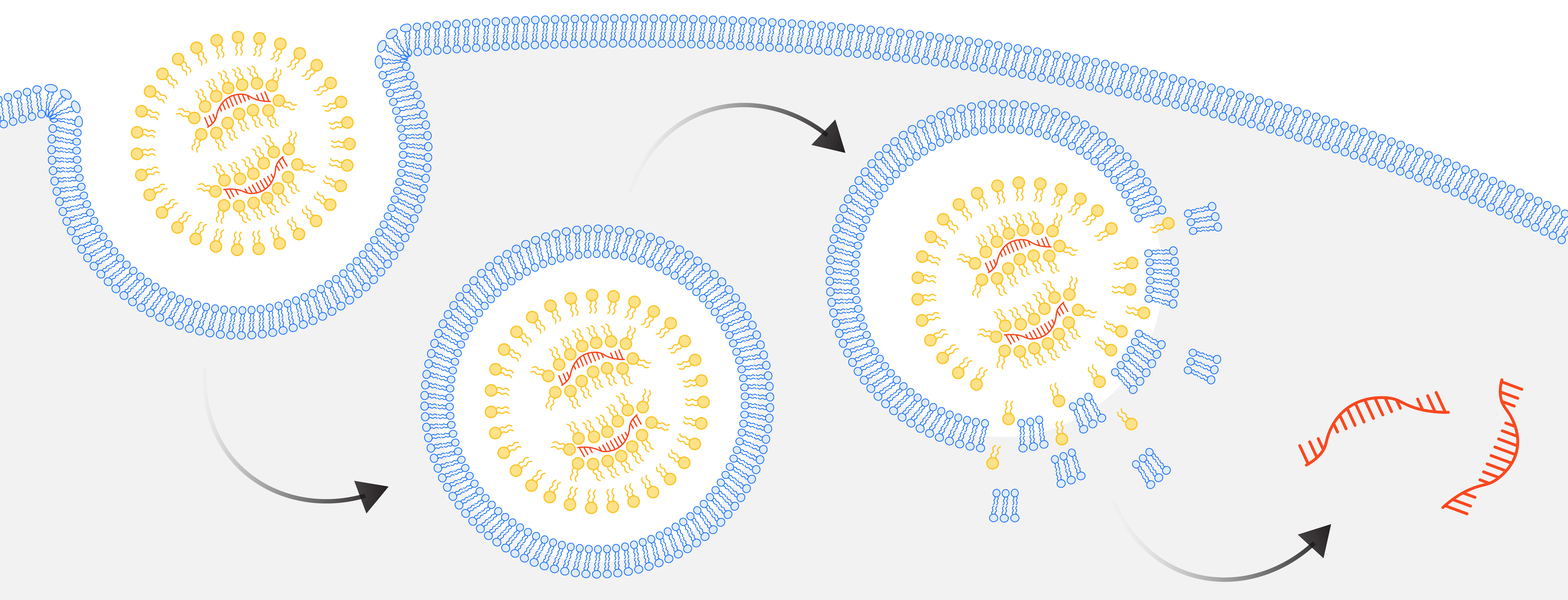
LNPs enter cells by endocytosis. To successfully deliver its cargo, an LNP must disrupt the endosome before it matures and breaks down its contents.
Next-generation viral vectors. While most FDA-approved genetic medicines rely on just two viral vectors, there are hundreds of thousands of viruses on Earth, including over 200 known to infect human cells. For example, Krystal Biotech's Vyjuvek is an HSV-1-based gene therapy approved in 2023 for treating the rare genetic skin disorder dystrophic epidermolysis bullosa (DEB).* Vyjuvek takes advantage of HSV’s inherent ability to avoid the immune system as well as its large cargo capacity to enable repeated delivery of a ~8,900 nucleotide collagen transcript to dividing skin cells. It remains to be seen whether HSV can be dosed systemically to treat disorders in other tissues. However, we imagine that the menagerie of viruses on Earth includes those with desirable properties like bioavailability in hard-to-reach tissues, low immunogenicity, and large cargo capacity.
In parallel, new technologies can enhance AAV and lentiviral vectors. The AAV capsid – the protein shell that encases its genome – can be engineered to include targeting moieties*, although this carries the risk of disrupting the capsid’s structural integrity. Tools for in vivo selection* can also be used to reveal capsids that are more efficient, target specific tissues, or are less immunogenic.
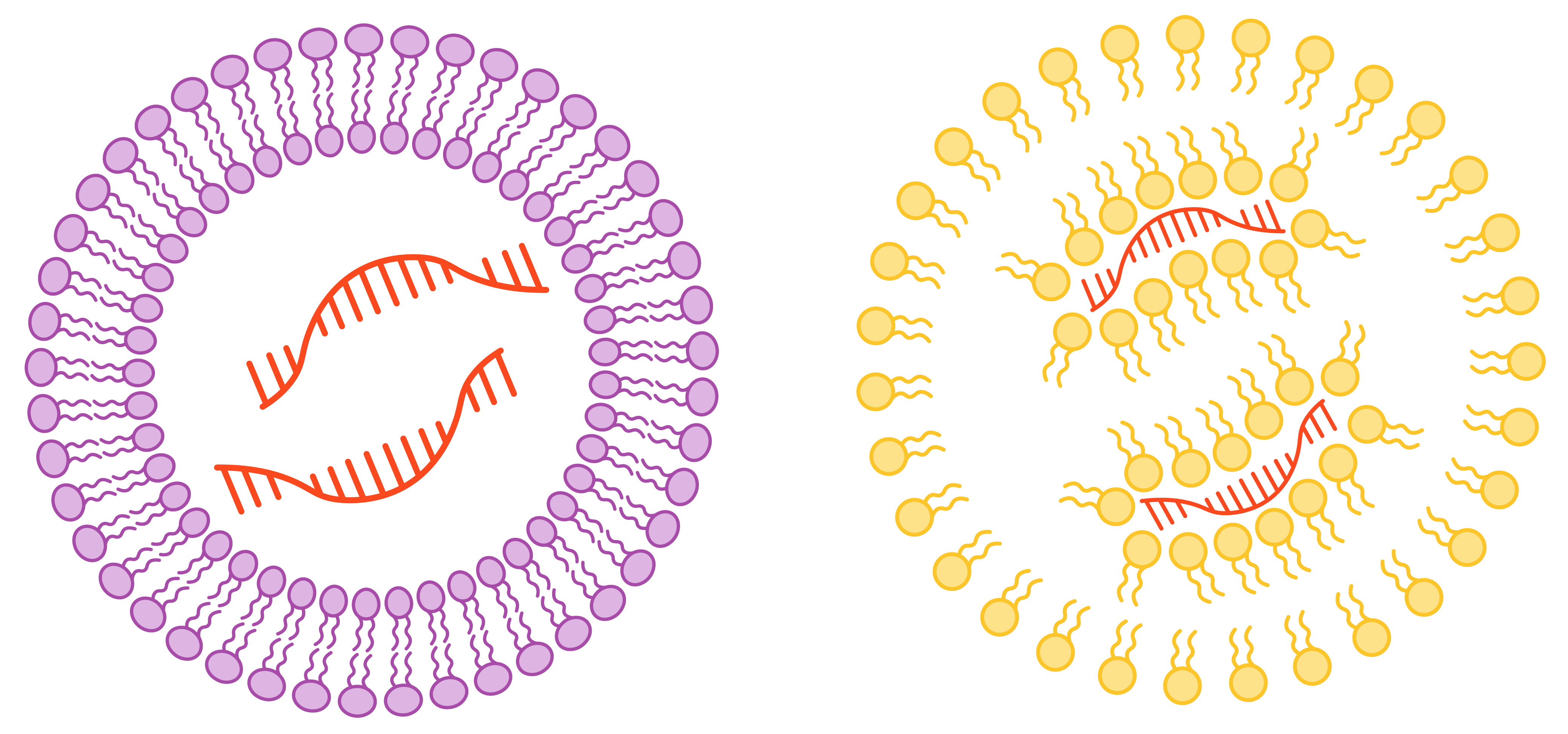
In contrast, the lentiviral capsid is surrounded by a lipid envelope that is more readily modified (psuedotyped) with targeting moieties or proteins that facilitate virus fusion with a cell membrane (fusogens).** Fusogens play a role in cell entry by mediating fusion of the viral envelope with the cell membrane (bypassing the endocytic pathway) or with the endosome (better facilitating endosomal escape). Certain fusogens may offer enhanced efficiency, exhibit natural specificity for certain cell types, or can be engineered for targeting specific cell types. However, certain modifications to the viral envelope can interfere with virus production.

Engineering the AAV capsid requires structural modifications to the capsid (left). In contrast, the lentiviral lipid envelope is easier to modify (right).
Extracellular vesicles (EVs). EVs are naturally occurring, lipid-bilayer-enclosed particles secreted from mammalian cells. They are thought to play a role in intercellular communication by transporting signals from cell to cell, suggesting that they could be used to deliver genetic material. EVs are already routinely administered during blood transfusions, as blood is naturally rich in EVs. This suggests that cultured EVs would be safe and non-immunogenic. However, they have short circulation half-lives, tend to accumulate in the liver, and their ability to reach tissues like the brain and bone marrow is controversial. This likely depends on the origin of the EVs: EVs produced by a certain cell type tend to home back to the same cell type, offering a potential strategy for targeted delivery. Like lentiviral vectors, EVs can also be engineered to express targeting moieties or fusogens on their surface.**
EVs share some limitations with viral vectors, including the need for expensive manufacturing processes using mammalian cells. Unlike viruses however, EVs do not generally package genetic material. This requires either that cells are engineered to efficiently load endogenous genetic material in EVs* or post-production exogenous loading (which can damage EV integrity). Similarly to LNPs, EVs are not thought to have a mechanism for entering the nucleus to deliver DNA to non-dividing cells. Additionally, EVs are inherently highly heterogeneous, often carrying additional cellular material such as proteins, metabolites, and small RNAs that might be undesirable for therapeutic applications.6 This makes production and isolation of uniform vesicles a challenge for scaling manufacturing.Viral-like particles (VLPs). VLPs are a hybrid between viruses and EVs. Like EVs, they are lipid-bilayer-enclosed particles secreted by cells. However, VLPs are engineered to incorporate viral components for enhancing secretion from cells, packaging genetic material, or facilitating cell entry. While they are considered safer than viruses because they lack viral genetic material, their viral components may be immunogenic. However, these viral components can be extensively engineered to reduce immunogenicity as well as optimize packaging and delivery**. Researchers have also used virus-like components derived from the human genome, which are presumably non-immunogenic*. It remains to be seen whether VLPs can avoid the liver and target specific tissues upon systemic delivery.
Synthetic liposomes. Synthetic liposomes combine advantageous features of both LNPs and EVs. Like LNPs, liposomes are chemically synthesized rather than produced by cells, which can allow for finer control over their composition and consistency. Like EVs, liposomes are lipid-bilayer-enclosed vesicles that can be conjugated with fusogens, allowing them to bypass or escape the endosome for enhanced efficiency. However, they also share certain limitations with LNPs and EVs, including potential toxicity, variable efficiency, and limited targeting of specific cell types. Their ability to enter the nucleus of non-dividing cells for DNA delivery is also controversial.

Synthetic liposomes (left) have a lipid bilayer, in contrast to LNPs (right).
The New Frontier: our investment in Nanite and its polymer nanoparticles (PNPs)
Towards overcoming the many limitations associated with existing gene delivery tools, Civilization portfolio company Nanite is developing polymer nanoparticles (PNPs) for gene delivery. The chief bottleneck for PNPs, however, is that their vast chemical diversity gives rise to millions of possible compositions, making it difficult to efficiently screen and identify optimal formulations for specific therapeutic applications.
The solution: Nanite has developed an AI-native platform, SAYER, that screens PNP formulations in silico and rapidly manufactures candidate PNPs. Nanite then evaluates PNPs both in cell lines and in vivo, taking advantage of technology that enables multiplexing dozens of different formulations in a single animal. Each design-build-test cycle is fed back into Nanite’s model, iteratively improving upon each PNP formulation and accelerating the development of future PNPs for new applications. Nanite’s PNPs fulfill many of our criteria for an ideal delivery vector:
Targetability. Nanite’s PNPs can target specific tissues and cell types while bypassing the liver, enabling precise gene expression and avoiding liver toxicity.
Delivery of large DNA cargo. Unlike LNPs that are largely limited to mRNA cargo and viral vectors that face strict cargo size limits, Nanite has demonstrated the delivery of large DNA cargo. This could enable the expression of large genes for the permanent and mutation-agnostic correction of a wide range of genetic disorders. Additionally, Nanite has demonstrated the repeat dosing of non-integrating DNA, opening the door to a variety of in situ and in vivo protein expression therapies. For other applications, Nanite’s PNPs can also be optimized for mRNA delivery.
Low immunogenicity. Nanite’s PNPs do not require viral proteins nor do they incorporate polyethylene glycol or ionizable lipids which are associated with immunogenicity to LNPs. This has the potential to prevent immune-related toxicities and could enable repeat dosing.
Inexpensive manufacturing. Nanite’s PNP manufacturing process is inexpensive and scalable. In contrast to the complexity and high costs associated with conjugating targeting proteins to LNPs, Nanite has also developed a scalable and consistent PNP bioconjugation process. This can enable the better targeting of specific cell types (e.g., immune cell subsets) without significantly driving up manufacturing costs.
As delivery continues to function as a bottleneck for the field of cell and gene therapy, we are excited to partner with the incredible team at Nanite to help unlock the full potential of these curative treatment modalities. Make sure to check out their recent preprint here.
While we focus here on gene therapies, we note that delivery is rate-limiting across biotech. New technologies are also needed for delivering small RNAs and protein drugs into cells and across biological barriers. On the flipside, new classes of small molecules can alter gene expression,*** with the potential to act like genetic medicines with far fewer delivery challenges.
As an additional complication, asymptomatic wild-type AAV infection is very common. Many prospective patients have pre-existing immunity that precludes them from receiving an AAV gene therapy.
While Sarepta recently received full approval for their muscle-targeted gene therapy, its efficacy is controversial.*
While viral vectors like AAV also can package random cytosolic materials, this is a bigger issue for EVs since they are more heterogenous and have mechanisms for packaging a wide variety of cytosolic cargo. In contrast, AAVs are smaller particles (limiting the amount of random material they can package) and use packaging signals to specifically encapsulate the intended genetic material.
 | A guest post by
|